¿Qué es un medicamento biosimilar?
Biosimilar: un biológico más
Un biosimilar (o medicamento biológico similar) es un medicamento biológico equivalente en calidad, eficacia y seguridad a un medicamento biológico original, llamado producto de referencia. La posología y vía de administración deben ser las mismas, y el biosimilar se autoriza para todas, o algunas de las indicaciones aprobadas para el biológico de referencia.
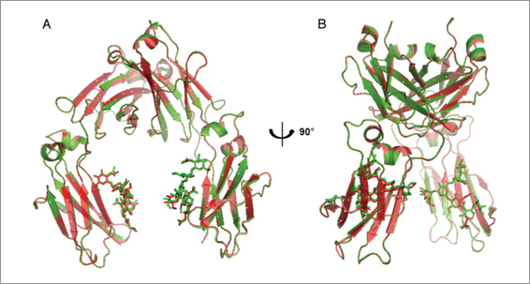
Superposición de estructuras cristalinas Fc CT-P13 (verde) y RMP Fc (rojo): (A) vista frontal; (B) vista lateral. Fuente: Physicochemical characterization of Remsima. Soon Kwan Junga, Kyoung Hoon Leea, Jae Won Jeona, Joon Won Leea, Byoung Oh Kwona, Yeon Jung Kima, Jin Soo Baea, Dong-Il Kimb, Soo Young Leea & Shin Jae Changa
El biosimilar contiene una versión del principio activo del producto de referencia. La equivalencia con este debe establecerse mediante un exhaustivo “ejercicio de comparabilidad” (del inglés “comparability exercise”). El objetivo de ese ejercicio es demostrar que las leves diferencias fisicoquímicas existentes entre ambos productos no inciden significativamente en el perfil beneficio/riesgo, lo cual permite sustentar que el principio activo de ambos medicamentos es en esencia el mismo. Una vez acreditada la equivalencia y autorizado, el biosimilar es un medicamento biológico más. La variabilidad fisicoquímica sin trascendencia terapéutica es inherente a productos de naturaleza biológica, e identificable en ocasiones entre lotes de determinados medicamentos biológicos sometidos a cambios en el proceso productivo. Lo cual puede exigir incluso la ejecución de un “ejercicio de comparabilidad” a la medida entre dos o más lotes de un mismo medicamento biológico, original o biosimilar, con el fin de comprobar su equivalencia.

Evidencia de eficacia/seguridad
La aprobación del biosimilar se sustenta sobre una rigurosa evidencia científico-médica, y las autoridades regulatorias aplican el mismo criterio en biológicos originales y similares (biosimilares) en relación al grado de exigencia en el equilibrio beneficio/riesgo estimado como aceptable.
La aprobación, o no, de un candidato a biosimilar depende del resultado del “ejercicio de comparabilidad”. La condición de biológicos de los productos comparados obliga a un ejercicio extenso y exhaustivo. La extensión y diseño del mismo dependerá del conocimiento clínico acumulado durante años de utilización terapéutica del medicamento de referencia, y de sus características farmacológicas. El “ejercicio de comparabilidad” por lo general abarca desde una detallada evaluación del grado de analogía estructural y funcional, hasta ensayos clínicos confirmatorios en pacientes. Lo cual supone entre 6 y 12 años de estudio. Su objetivo es acreditar con suficientes garantías que ambos productos comparten eficacia y perfil de seguridad.
La aprobación, o no, de un candidato a biosimilar depende del resultado del “ejercicio de comparabilidad”. La condición de biológicos de los productos comparados obliga a un ejercicio extenso y exhaustivo. La extensión y diseño del mismo dependerá del conocimiento clínico acumulado durante años de utilización terapéutica del medicamento de referencia, y de sus características farmacológicas. El “ejercicio de comparabilidad” por lo general abarca desde una detallada evaluación del grado de analogía estructural y funcional, hasta ensayos clínicos confirmatorios en pacientes. Lo cual supone entre 6 y 12 años de estudio. Su objetivo es acreditar con suficientes garantías que ambos productos comparten eficacia y perfil de seguridad.
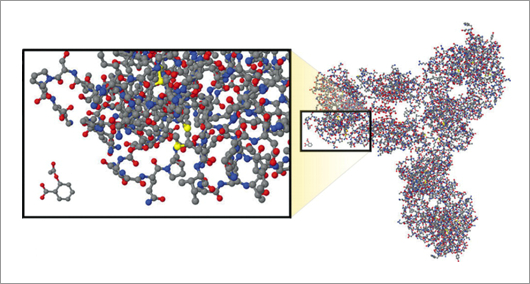
Comparativa entre un anticuerpo monoclonal biológico y una molécula de ácido acetilsalicílico. Fuente: Developing the Nation’s Biosimilars Program. Steven Kozlowski, M.D., Janet Woodcock, M.D., Karen Midthun, M.D., and Rachel Behrman Sherman, M.D., M.P.H.
La demostración de equivalencia entre medicamentos que no tiene un origen biológico, como un candidato a genérico y el producto original de síntesis química (o «molécula pequeña») con el que se compara, no requiere de una evaluación tan extensa y exhaustiva puesto que es posible generar principios activos de síntesis química virtualmente idénticos en su estructura. Si bien la base legal sobre la que se asientan es equiparable, no deben confundirse los conceptos biosimilar y genérico. De igual manera, no es científicamente aconsejable hablar de biogenéricos.

Garantía EMA
La autorización de medicamentos biosimilares, de origen biotecnológico, en los países del Área Económica Europea (AEE), como España, está sujeta a un procedimiento centralizado. Es decir que es la Agencia Europea del Medicamento (EMA), ubicada en Ámsterdam, la institución responsable de evaluar el dossier (o expediente) que recoge los estudios realizados con el candidato a biosimilar, y emitir un informe respecto a la acreditación, o no, de biosimilitud. Los evaluadores de la EMA son expertos que pertenecen a las agencias reguladoras de cada uno de los estados miembros.
La EMA fue pionera en elaborar directrices (guidelines) acerca de cómo deben desarrollarse los biosimilares; es decir, acerca de los estudios requeridos para demostrar la equivalencia. Los criterios de ese marco regulatorio fueron luego reproducidos en su práctica literalidad por la Organización Mundial de la Salud (OMS), y recogidos en esencia por agencias reguladoras de referencia (la estadounidense FDA, la canadiense, la japonesa y la australiana). El equipo de expertos que evalúa los expedientes de los medicamentos biológicos originales también lo hace con los candidatos a biosimilar, y necesariamente aplica el mismo criterio respecto a la exigible garantía de eficacia y seguridad. Por lo tanto, la pertenencia al AEE tiene un especial significado en el terreno de los biosimilares, por el muy particular liderazgo y reconocimiento que se atribuye a la EMA respecto a la experiencia en los requerimientos regulatorios exigibles, y en la evaluación pre-comercial de estos productos.

Accesibilidad de pacientes
El principal, pero no único, valor añadido atribuible a la entrada de los biosimilares es la posibilidad de que un mayor número de pacientes acceda a tratamientos biológicos. Es decir que, debido a la reducción del coste respecto al producto original, el principal beneficiario de la incorporación de los medicamentos biosimilares a los sistemas sanitarios son los pacientes.

Sostenibilidad del sistema
Los medicamentos biológicos han supuesto un notable avance en el tratamiento de enfermedades por lo general graves y crónicas (pero no únicamente). Se trata ya en muchos casos de medicamentos terapéuticamente esenciales, pero cuyo coste medio es muy superior al de los medicamentos de síntesis química. Pueden llegar a requerir en determinados hospitales más del 40% de los recursos farmacéuticos. Es una tendencia al alza, porque se sabe que un tanto por ciento elevado de los productos actualmente en desarrollo clínico son biotecnológicos. Los biosimilares, productos biológicos equivalente pero con menor precio, contribuyen al ahorro en la factura farmacéutica sin reducir el acceso a terapias biológicas de calidad. Los biosimilares consolidan por lo tanto la garantía de una cobertura sanitaria de calidad para futuras generaciones de pacientes.

Acceso a productos innovadores
La incorporación de los medicamentos biosimilares contribuirá a acrecentar sustancialmente el acceso de los pacientes a tratamientos innovadores. Este mayor acceso se produce por dos mecanismos. Por un lado, el ahorro hospitalario que supone la adquisición de terapias biológicas menos costosas, puede reconducirse hacia nuevas opciones terapéuticas biológicas que no son costeables hoy en día para los pacientes que puedan requerirlas. Por otro, al promover la competencia en el mercado farmacéutico, los medicamentos biosimilares contribuyen a incentivar la investigación de nuevos productos por parte de la industria biofarmacéutica.